My Python notebook: displaying the state of the Genome Projects
This post if about my first Python program. I'm still a newbie so please, don't flame ! :-)
The NCBI hosts a XML file describing the state of the Genome Projects at ftp://ftp.ncbi.nih.gov/genomes/genomeprj/gp.xml. The very first lines of the file look like this:
<?xml version="1.0" encoding="UTF-8"?>
<!DOCTYPE gp:DocumentSet SYSTEM "gp4v.dtd">
<gp:DocumentSet xmlns:SOAP-ENV="http://schemas.xmlsoap.org/soap/envelope/" xmlns:SOAP-ENC="http://s
chemas.xmlsoap.org/soap/encoding/" xmlns:xsi="http://www.w3.org/2001/XMLSchema-instance" xmlns:xsd="http://www.w3.org/2001/XMLSchema" xmlns:gp="gp">
<gp:Document>
<gp:ProjectID>1</gp:ProjectID>
<gp:IsPrivate>false</gp:IsPrivate>
<gp:eType>eGenome</gp:eType>
<gp:eMethod>eWGS</gp:eMethod>
<gp:eTechnologies>eUnspecified</gp:eTechnologies>
<gp:OriginDB>NCBI</gp:OriginDB>
<gp:CreateDate>03/05/2003 15:03:16</gp:CreateDate>
<gp:ModificationDate>07/18/2006 16:09:02</gp:ModificationDate>
<gp:ProjectName/>
<gp:OrganismName>Acidobacterium capsulatum ATCC 51196</gp:OrganismName>
<gp:StrainName>ATCC 51196</gp:StrainName>
<gp:TaxID>240015</gp:TaxID>
<gp:LocusTagPrefix/>
<gp:DNASource/>
<gp:SequencingDepth/>
<gp:ProjectURL>http://www.tigr.org/tdb/mdb/mdbinprogress.html</gp:ProjectURL>
<gp:DataURL/>
<gp:OrganismDescription/>
<gp:EstimatedGenomeSize>4.15</gp:EstimatedGenomeSize>
(...)
<gp:ChromosomeDefinitions/>
<gp:SubmittedSequences/>
<gp:LegacyPrefixes/>
</gp:Document>
<gp:Document>
<gp:ProjectID>3</gp:ProjectID>
<gp:IsPrivate>false</gp:IsPrivate>
<gp:eType>eGenome</gp:eType>
(...)
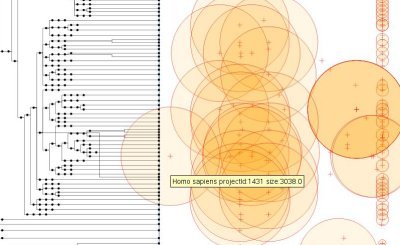
The program I wrote reads this XML file and generates the following visualization:<!DOCTYPE gp:DocumentSet SYSTEM "gp4v.dtd">
<gp:DocumentSet xmlns:SOAP-ENV="http://schemas.xmlsoap.org/soap/envelope/" xmlns:SOAP-ENC="http://s
chemas.xmlsoap.org/soap/encoding/" xmlns:xsi="http://www.w3.org/2001/XMLSchema-instance" xmlns:xsd="http://www.w3.org/2001/XMLSchema" xmlns:gp="gp">
<gp:Document>
<gp:ProjectID>1</gp:ProjectID>
<gp:IsPrivate>false</gp:IsPrivate>
<gp:eType>eGenome</gp:eType>
<gp:eMethod>eWGS</gp:eMethod>
<gp:eTechnologies>eUnspecified</gp:eTechnologies>
<gp:OriginDB>NCBI</gp:OriginDB>
<gp:CreateDate>03/05/2003 15:03:16</gp:CreateDate>
<gp:ModificationDate>07/18/2006 16:09:02</gp:ModificationDate>
<gp:ProjectName/>
<gp:OrganismName>Acidobacterium capsulatum ATCC 51196</gp:OrganismName>
<gp:StrainName>ATCC 51196</gp:StrainName>
<gp:TaxID>240015</gp:TaxID>
<gp:LocusTagPrefix/>
<gp:DNASource/>
<gp:SequencingDepth/>
<gp:ProjectURL>http://www.tigr.org/tdb/mdb/mdbinprogress.html</gp:ProjectURL>
<gp:DataURL/>
<gp:OrganismDescription/>
<gp:EstimatedGenomeSize>4.15</gp:EstimatedGenomeSize>
(...)
<gp:ChromosomeDefinitions/>
<gp:SubmittedSequences/>
<gp:LegacyPrefixes/>
</gp:Document>
<gp:Document>
<gp:ProjectID>3</gp:ProjectID>
<gp:IsPrivate>false</gp:IsPrivate>
<gp:eType>eGenome</gp:eType>
(...)
There are two main classes: A Taxon is a node in the NCBI taxonomy, it contains a link to his children and to his parent:
class Taxon:
def __init__(self):
self.id=-1
self.name=None
self.parent=None
self.children=set()
. A Project is an item in the XML file. It contains a link to a Taxon:def __init__(self):
self.id=-1
self.name=None
self.parent=None
self.children=set()
class Project:
def __init__(self):
self.id=-1
self.taxon=None
self.genomeSize=None
self.date=None
The XML file is processed with a SAX handler. Each time a new project is found, the corresponding taxon is downloaded :def __init__(self):
self.id=-1
self.taxon=None
self.genomeSize=None
self.date=None
class GPHandler(handler.ContentHandler):
LIMIT=1E7
SLEEP=0
def __init__(self,owner):
self.owner=owner
self.content=None
self.project=None
(...)
def startElementNS(self,name,qname,attrs):
if name[1]=="Document":
self.project=Project()
elif name[1] in ["ProjectID","TaxID","CreateDate","EstimatedGenomeSize"] :
self.content=""
def endElementNS(self,name,qname):
if name[1]=="Document":
if len(self.owner.id2project)<GPHandler.LIMIT:
self.owner.id2project[self.project.id]= self.project
self.project=None
elif name[1]=="TaxID":
if len(self.owner.id2project)<GPHandler.LIMIT:
self.project.taxon=self.owner.findTaxonById(int(self.content))
time.sleep(GPHandler.SLEEP) #don't be evil with NCBI
elif name[1]=="ProjectID":
self.project.id=int(self.content)
sys.stderr.write("Project id:"+self.content+" n="+str(len(self.owner.id2project))+"\n")
elif name[1]=="EstimatedGenomeSize" and len(self.content)>0:
self.project.genomeSize=float(self.content)
elif name[1]=="CreateDate":
self.project.date=self.sql2date(self.content)
self.content=None
def characters(self,content):
if self.content!=None:
self.content+=content
The XML description of each taxons is downloaded and parsed using the NCBI EFetch service. A XML for a taxon looks like this:LIMIT=1E7
SLEEP=0
def __init__(self,owner):
self.owner=owner
self.content=None
self.project=None
(...)
def startElementNS(self,name,qname,attrs):
if name[1]=="Document":
self.project=Project()
elif name[1] in ["ProjectID","TaxID","CreateDate","EstimatedGenomeSize"] :
self.content=""
def endElementNS(self,name,qname):
if name[1]=="Document":
if len(self.owner.id2project)<GPHandler.LIMIT:
self.owner.id2project[self.project.id]= self.project
self.project=None
elif name[1]=="TaxID":
if len(self.owner.id2project)<GPHandler.LIMIT:
self.project.taxon=self.owner.findTaxonById(int(self.content))
time.sleep(GPHandler.SLEEP) #don't be evil with NCBI
elif name[1]=="ProjectID":
self.project.id=int(self.content)
sys.stderr.write("Project id:"+self.content+" n="+str(len(self.owner.id2project))+"\n")
elif name[1]=="EstimatedGenomeSize" and len(self.content)>0:
self.project.genomeSize=float(self.content)
elif name[1]=="CreateDate":
self.project.date=self.sql2date(self.content)
self.content=None
def characters(self,content):
if self.content!=None:
self.content+=content
<TaxaSet>
<Taxon>
<TaxId>240015</TaxId>
<ScientificName>Acidobacterium capsulatum ATCC 51196</ScientificName>
<OtherNames>
<EquivalentName>Acidobacterium capsulatum strain ATCC 51196</EquivalentName>
<EquivalentName>Acidobacterium capsulatum str. ATCC 51196</EquivalentName>
</OtherNames>
<ParentTaxId>33075</ParentTaxId>
<Rank>no rank</Rank>
<Division>Bacteria</Division>
<GeneticCode>
<GCId>11</GCId>
<GCName>Bacterial, Archaeal and Plant Plastid</GCName>
</GeneticCode>
<MitoGeneticCode>
<MGCId>0</MGCId>
<MGCName>Unspecified</MGCName>
</MitoGeneticCode>
<Lineage>cellular organisms; Bacteria; Fibrobacteres/Acidobacteria group; Acidobacteria; Acidobacteria (class); Acidobacteriales; Acidobacteriaceae; Acidobacterium; Acidobacterium capsulatum</Lineage>
<LineageEx>
<Taxon>
<TaxId>131567</TaxId>
<ScientificName>cellular organisms</ScientificName>
<Rank>no rank</Rank>
</Taxon>
<Taxon>
<TaxId>2</TaxId>
<ScientificName>Bacteria</ScientificName>
<Rank>superkingdom</Rank>
</Taxon>
<Taxon>
<TaxId>131550</TaxId>
<ScientificName>Fibrobacteres/Acidobacteria group</ScientificName>
<Rank>superphylum</Rank>
</Taxon>
<Taxon>
<TaxId>57723</TaxId>
<ScientificName>Acidobacteria</ScientificName>
<Rank>phylum</Rank>
</Taxon>
<Taxon>
<TaxId>204432</TaxId>
<ScientificName>Acidobacteria (class)</ScientificName>
<Rank>class</Rank>
</Taxon>
<Taxon>
<TaxId>204433</TaxId>
<ScientificName>Acidobacteriales</ScientificName>
<Rank>order</Rank>
</Taxon>
<Taxon>
<TaxId>204434</TaxId>
<ScientificName>Acidobacteriaceae</ScientificName>
<Rank>family</Rank>
</Taxon>
<Taxon>
<TaxId>33973</TaxId>
<ScientificName>Acidobacterium</ScientificName>
<Rank>genus</Rank>
</Taxon>
<Taxon>
<TaxId>33075</TaxId>
<ScientificName>Acidobacterium capsulatum</ScientificName>
<Rank>species</Rank>
</Taxon>
</LineageEx>
<CreateDate>2003/07/25</CreateDate>
<UpdateDate>2005/01/19</UpdateDate>
<PubDate>2005/01/29</PubDate>
</Taxon>
</TaxaSet>
This time I used a DOM implementation for python to analyse the file:<Taxon>
<TaxId>240015</TaxId>
<ScientificName>Acidobacterium capsulatum ATCC 51196</ScientificName>
<OtherNames>
<EquivalentName>Acidobacterium capsulatum strain ATCC 51196</EquivalentName>
<EquivalentName>Acidobacterium capsulatum str. ATCC 51196</EquivalentName>
</OtherNames>
<ParentTaxId>33075</ParentTaxId>
<Rank>no rank</Rank>
<Division>Bacteria</Division>
<GeneticCode>
<GCId>11</GCId>
<GCName>Bacterial, Archaeal and Plant Plastid</GCName>
</GeneticCode>
<MitoGeneticCode>
<MGCId>0</MGCId>
<MGCName>Unspecified</MGCName>
</MitoGeneticCode>
<Lineage>cellular organisms; Bacteria; Fibrobacteres/Acidobacteria group; Acidobacteria; Acidobacteria (class); Acidobacteriales; Acidobacteriaceae; Acidobacterium; Acidobacterium capsulatum</Lineage>
<LineageEx>
<Taxon>
<TaxId>131567</TaxId>
<ScientificName>cellular organisms</ScientificName>
<Rank>no rank</Rank>
</Taxon>
<Taxon>
<TaxId>2</TaxId>
<ScientificName>Bacteria</ScientificName>
<Rank>superkingdom</Rank>
</Taxon>
<Taxon>
<TaxId>131550</TaxId>
<ScientificName>Fibrobacteres/Acidobacteria group</ScientificName>
<Rank>superphylum</Rank>
</Taxon>
<Taxon>
<TaxId>57723</TaxId>
<ScientificName>Acidobacteria</ScientificName>
<Rank>phylum</Rank>
</Taxon>
<Taxon>
<TaxId>204432</TaxId>
<ScientificName>Acidobacteria (class)</ScientificName>
<Rank>class</Rank>
</Taxon>
<Taxon>
<TaxId>204433</TaxId>
<ScientificName>Acidobacteriales</ScientificName>
<Rank>order</Rank>
</Taxon>
<Taxon>
<TaxId>204434</TaxId>
<ScientificName>Acidobacteriaceae</ScientificName>
<Rank>family</Rank>
</Taxon>
<Taxon>
<TaxId>33973</TaxId>
<ScientificName>Acidobacterium</ScientificName>
<Rank>genus</Rank>
</Taxon>
<Taxon>
<TaxId>33075</TaxId>
<ScientificName>Acidobacterium capsulatum</ScientificName>
<Rank>species</Rank>
</Taxon>
</LineageEx>
<CreateDate>2003/07/25</CreateDate>
<UpdateDate>2005/01/19</UpdateDate>
<PubDate>2005/01/29</PubDate>
</Taxon>
</TaxaSet>
def findTaxonById(self,taxId):
if taxId in self.id2taxon:
return self.id2taxon[taxId]
url="http://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=taxonomy&retmode=xml&id="+str(taxId)
new_taxon=Taxon()
new_taxon.id=taxId
new_taxon.parent
dom = xml.dom.minidom.parse(urllib.urlopen(url))
top= dom.documentElement
e1= self.first(top,"Taxon")
new_taxon.name = self.textContent(self.first(e1,"ScientificName"))
lineage=[]
lineage.append(self.taxonRoot)
for e2 in self.elements(self.first(e1,"LineageEx"),"Taxon"):
t2=Taxon()
t2.id= int(self.textContent(self.first(e2,"TaxId")))
t2.name= self.textContent(self.first(e2,"ScientificName"))
if t2.id in self.id2taxon:
t2= self.id2taxon[t2.id]
else:
self.id2taxon[t2.id]=t2
lineage.append(t2)
lineage.append(new_taxon)
i=1
while i < len(lineage):
lineage[i-1].children.add(lineage[i])
lineage[i].parent=lineage[i-1]
i+=1
self.id2taxon[new_taxon.id]=new_taxon
return new_taxon
At the end, the figure is generated using SVG:if taxId in self.id2taxon:
return self.id2taxon[taxId]
url="http://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=taxonomy&retmode=xml&id="+str(taxId)
new_taxon=Taxon()
new_taxon.id=taxId
new_taxon.parent
dom = xml.dom.minidom.parse(urllib.urlopen(url))
top= dom.documentElement
e1= self.first(top,"Taxon")
new_taxon.name = self.textContent(self.first(e1,"ScientificName"))
lineage=[]
lineage.append(self.taxonRoot)
for e2 in self.elements(self.first(e1,"LineageEx"),"Taxon"):
t2=Taxon()
t2.id= int(self.textContent(self.first(e2,"TaxId")))
t2.name= self.textContent(self.first(e2,"ScientificName"))
if t2.id in self.id2taxon:
t2= self.id2taxon[t2.id]
else:
self.id2taxon[t2.id]=t2
lineage.append(t2)
lineage.append(new_taxon)
i=1
while i < len(lineage):
lineage[i-1].children.add(lineage[i])
lineage[i].parent=lineage[i-1]
i+=1
self.id2taxon[new_taxon.id]=new_taxon
return new_taxon
Source code
#Author Pierre Lindenbaum
#Mail: plindenbaum@yahoo.fr
#WWW: http://plindenbaum.blogspot.com
#usage:
# wget -O gp.xml "ftp://ftp.ncbi.nih.gov/genomes/genomeprj/gp.xml"
# python gp.py gp.xml
from xml.sax import make_parser, handler,saxutils
import sys,time,math,pickle,os.path
import xml.dom.minidom
import urllib
class SVG:
NS="http://www.w3.org/2000/svg"
class XLINK:
NS="http://www.w3.org/1999/xlink"
class Main:
DB_FILE="gp.db"
def __init__(self):
self.id2project=dict()
self.id2taxon=dict()
self.min_size=1.0E13
self.max_size=0.0
self.min_date=1.0E13
self.max_date=0.0
self.taxonRoot=Taxon()
self.taxonRoot.id=0
self.taxonRoot.name="Tree of Life"
self.id2taxon[self.taxonRoot.id]=self.taxonRoot
def escape(self,s):
x=""
for c in s:
if c=='<':
x+="<"
elif c=='>':
x+=">"
elif c=='&':
x+="&"
elif c=='\'':
x+="'"
elif c=='\'':
x+="""
else:
x+=c
return x
def first(self,root,name):
for c1 in root.childNodes:
if c1.nodeType!= xml.dom.minidom.Node.ELEMENT_NODE or \
c1.nodeName!=name:
continue
return c1
return None
def elements(self,root,name):
a=[]
for c1 in root.childNodes:
if c1.nodeType!= xml.dom.minidom.Node.ELEMENT_NODE or \
c1.nodeName!=name:
continue
a.append(c1)
return a
def textContent(self,root):
content=""
for c1 in root.childNodes:
if c1.nodeType== xml.dom.minidom.Node.TEXT_NODE:
content+= c1.nodeValue
else:
content+=self.textContent(c1)
return content
def findTaxonById(self,taxId):
if taxId in self.id2taxon:
return self.id2taxon[taxId]
url="http://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=taxonomy&retmode=xml&id="+str(taxId)
sys.stderr.write(url+"\n")
new_taxon=Taxon()
new_taxon.id=taxId
new_taxon.parent
dom = xml.dom.minidom.parse(urllib.urlopen(url))
top= dom.documentElement
e1= self.first(top,"Taxon")
new_taxon.name = self.textContent(self.first(e1,"ScientificName"))
lineage=[]
lineage.append(self.taxonRoot)
for e2 in self.elements(self.first(e1,"LineageEx"),"Taxon"):
t2=Taxon()
t2.id= int(self.textContent(self.first(e2,"TaxId")))
t2.name= self.textContent(self.first(e2,"ScientificName"))
if t2.id in self.id2taxon:
t2= self.id2taxon[t2.id]
else:
self.id2taxon[t2.id]=t2
lineage.append(t2)
lineage.append(new_taxon)
i=1
while i < len(lineage):
lineage[i-1].children.add(lineage[i])
lineage[i].parent=lineage[i-1]
i+=1
self.id2taxon[new_taxon.id]=new_taxon
return new_taxon
def svg(self):
width= 1000
height=10000
for p in self.id2project.values():
if p.genomeSize!=None:
self.min_size = min(self.min_size, p.genomeSize)
self.max_size = max(self.max_size, p.genomeSize)
self.min_date = min(self.min_date, p.date)
self.max_date = max(self.max_date, p.date)
shift= (self.max_date-self.min_date)*0.05
self.min_date-=shift
self.max_date+=shift
sys.stderr.write(" size:"+str(self.min_size)+"/"+str(self.max_size))
sys.stderr.write(" date:"+str(self.min_date)+"/"+str(self.max_date))
print "<?xml version='1.0' encoding='UTF-8'?>"
print "<svg xmlns='"+SVG.NS+"' xmlns:xlink='"+XLINK.NS+"' "+\
"width='"+str(width+1)+"' "+\
"height='"+str(height+1)+"' "+\
"style='stroke:black;fill:none;stroke-width:0.5px;' "+\
"><title>Genome Projects</title>"
print "<rect x='0' y='0' width='"+str(width/2.0)+"' height='"+str(height)+"' style='stroke:green;'/>"
print "<rect x='"+str(1+width/2.0)+"' y='0' width='"+str(width/2.0)+"' height='"+str(height)+"' style='stroke:blue;'/>"
self.recurs(self.taxonRoot,0,0,width/2,height,(width/2.0)/self.taxonRoot.depth())
print "</svg>"
def recurs(self,taxon,x,y,width,height,h_size):
total=0.0
midy=y+height/2.0
print "<circle cx='"+str(x)+"' cy='"+str(midy)+"' r='"+str(2)+"' title='"+self.escape(taxon.name)+"' style='fill:black;'/>"
for p in self.id2project.values():
if p.taxon!=taxon:
continue
cx= width+ ((p.date - self.min_date)/(self.max_date - self.min_date))*width
cy= midy
print "<a target='ncbi' title='"+self.escape(taxon.name)+" projectId:"+str(p.id)+" size:"+str(p.genomeSize)+"' xlink:href='http://www.ncbi.nlm.nih.gov/sites/entrez?Db=genomeprj&cmd=ShowDetailView&TermToSearch="+str(p.id)+"'>"
print "<g style='stroke:red;' >"
if p.genomeSize!=None:
radius=max(2.0,((math.log10(p.genomeSize) - math.log10(self.min_size))/(math.log10(self.max_size) - math.log10(self.min_size)))*100.0)
print "<circle cx='"+str(cx)+"' cy='"+str(cy)+"' r='"+str(radius)+"' style='fill:orange;fill-opacity:0.1;'/>"
print "<line x1='"+str(cx-5)+"' y1='"+str(cy)+"' x2='"+str(cx+5)+"' y2='"+str(cy)+"'/>"
print "<line x1='"+str(cx)+"' y1='"+str(cy-5)+"' x2='"+str(cx)+"' y2='"+str(cy+5)+"'/>"
print "</g></a>"
for c in taxon.children:
total+= c.weight()
for c in taxon.children:
h2=height* (c.weight()/total)
y2=y+h2/2.0
w2=h_size
if len(c.children)==0:
w2=width-x
print "<polyline points='"+\
str(x)+","+str(midy)+" "+\
str(x+w2/2.0)+","+str(midy)+" "+\
str(x+w2/2.0)+","+str(y2)+" "+\
str(x+w2)+","+str(y2)+" "+\
"' title='"+self.escape(c.name)+"'/>"
self.recurs(c,x+w2,y,width,h2,w2)
y+=h2
class Taxon:
def __init__(self):
self.id=-1
self.name=None
self.parent=None
self.children=set()
def __hash__(self):
return self.id.__hash__()
def __eq__(self,other):
if other==None:
return False
return self.id==other.id
def weight(self):
w=1
for t in self.children:
w+= t.weight()
return w
def depth(self):
##sys.stderr.write(self.name+"\n")
d=0.0
for t in self.children:
d= max(t.depth(),d)
return 1.0+d
class Project:
def __init__(self):
self.id=-1
self.taxon=None
self.genomeSize=None
self.date=None
class GPHandler(handler.ContentHandler):
LIMIT=1E7
SLEEP=0
def __init__(self,owner):
self.owner=owner
self.content=None
self.project=None
def sql2date(self,s):
s=s[:s.index(' ')]
cal =time.mktime( time.strptime(s,"%m/%d/%Y") )
return cal
def startElementNS(self,name,qname,attrs):
if name[1]=="Document":
self.project=Project()
elif name[1] in ["ProjectID","TaxID","CreateDate","EstimatedGenomeSize"] :
self.content=""
def endElementNS(self,name,qname):
if name[1]=="Document":
if len(self.owner.id2project)<GPHandler.LIMIT:
self.owner.id2project[self.project.id]= self.project
self.project=None
elif name[1]=="TaxID":
if len(self.owner.id2project)<GPHandler.LIMIT:
self.project.taxon=self.owner.findTaxonById(int(self.content))
time.sleep(GPHandler.SLEEP) #don't be evil with NCBI
elif name[1]=="ProjectID":
self.project.id=int(self.content)
sys.stderr.write("Project id:"+self.content+" n="+str(len(self.owner.id2project))+"\n")
elif name[1]=="EstimatedGenomeSize" and len(self.content)>0:
self.project.genomeSize=float(self.content)
elif name[1]=="CreateDate":
self.project.date=self.sql2date(self.content)
self.content=None
def characters(self,content):
if self.content!=None:
self.content+=content
main=Main();
sys.stderr.write(main.escape("<>!")+"\n")
parser = make_parser()
# we want XML namespaces
parser.setFeature(handler.feature_namespaces,True)
# tell parser to disable validation
parser.setFeature(handler.feature_validation,False)
parser.setFeature(handler.feature_external_ges, False)
parser.setContentHandler(GPHandler(main))
parser.parse(sys.argv[1])
main.svg()
#Mail: plindenbaum@yahoo.fr
#WWW: http://plindenbaum.blogspot.com
#usage:
# wget -O gp.xml "ftp://ftp.ncbi.nih.gov/genomes/genomeprj/gp.xml"
# python gp.py gp.xml
from xml.sax import make_parser, handler,saxutils
import sys,time,math,pickle,os.path
import xml.dom.minidom
import urllib
class SVG:
NS="http://www.w3.org/2000/svg"
class XLINK:
NS="http://www.w3.org/1999/xlink"
class Main:
DB_FILE="gp.db"
def __init__(self):
self.id2project=dict()
self.id2taxon=dict()
self.min_size=1.0E13
self.max_size=0.0
self.min_date=1.0E13
self.max_date=0.0
self.taxonRoot=Taxon()
self.taxonRoot.id=0
self.taxonRoot.name="Tree of Life"
self.id2taxon[self.taxonRoot.id]=self.taxonRoot
def escape(self,s):
x=""
for c in s:
if c=='<':
x+="<"
elif c=='>':
x+=">"
elif c=='&':
x+="&"
elif c=='\'':
x+="'"
elif c=='\'':
x+="""
else:
x+=c
return x
def first(self,root,name):
for c1 in root.childNodes:
if c1.nodeType!= xml.dom.minidom.Node.ELEMENT_NODE or \
c1.nodeName!=name:
continue
return c1
return None
def elements(self,root,name):
a=[]
for c1 in root.childNodes:
if c1.nodeType!= xml.dom.minidom.Node.ELEMENT_NODE or \
c1.nodeName!=name:
continue
a.append(c1)
return a
def textContent(self,root):
content=""
for c1 in root.childNodes:
if c1.nodeType== xml.dom.minidom.Node.TEXT_NODE:
content+= c1.nodeValue
else:
content+=self.textContent(c1)
return content
def findTaxonById(self,taxId):
if taxId in self.id2taxon:
return self.id2taxon[taxId]
url="http://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=taxonomy&retmode=xml&id="+str(taxId)
sys.stderr.write(url+"\n")
new_taxon=Taxon()
new_taxon.id=taxId
new_taxon.parent
dom = xml.dom.minidom.parse(urllib.urlopen(url))
top= dom.documentElement
e1= self.first(top,"Taxon")
new_taxon.name = self.textContent(self.first(e1,"ScientificName"))
lineage=[]
lineage.append(self.taxonRoot)
for e2 in self.elements(self.first(e1,"LineageEx"),"Taxon"):
t2=Taxon()
t2.id= int(self.textContent(self.first(e2,"TaxId")))
t2.name= self.textContent(self.first(e2,"ScientificName"))
if t2.id in self.id2taxon:
t2= self.id2taxon[t2.id]
else:
self.id2taxon[t2.id]=t2
lineage.append(t2)
lineage.append(new_taxon)
i=1
while i < len(lineage):
lineage[i-1].children.add(lineage[i])
lineage[i].parent=lineage[i-1]
i+=1
self.id2taxon[new_taxon.id]=new_taxon
return new_taxon
def svg(self):
width= 1000
height=10000
for p in self.id2project.values():
if p.genomeSize!=None:
self.min_size = min(self.min_size, p.genomeSize)
self.max_size = max(self.max_size, p.genomeSize)
self.min_date = min(self.min_date, p.date)
self.max_date = max(self.max_date, p.date)
shift= (self.max_date-self.min_date)*0.05
self.min_date-=shift
self.max_date+=shift
sys.stderr.write(" size:"+str(self.min_size)+"/"+str(self.max_size))
sys.stderr.write(" date:"+str(self.min_date)+"/"+str(self.max_date))
print "<?xml version='1.0' encoding='UTF-8'?>"
print "<svg xmlns='"+SVG.NS+"' xmlns:xlink='"+XLINK.NS+"' "+\
"width='"+str(width+1)+"' "+\
"height='"+str(height+1)+"' "+\
"style='stroke:black;fill:none;stroke-width:0.5px;' "+\
"><title>Genome Projects</title>"
print "<rect x='0' y='0' width='"+str(width/2.0)+"' height='"+str(height)+"' style='stroke:green;'/>"
print "<rect x='"+str(1+width/2.0)+"' y='0' width='"+str(width/2.0)+"' height='"+str(height)+"' style='stroke:blue;'/>"
self.recurs(self.taxonRoot,0,0,width/2,height,(width/2.0)/self.taxonRoot.depth())
print "</svg>"
def recurs(self,taxon,x,y,width,height,h_size):
total=0.0
midy=y+height/2.0
print "<circle cx='"+str(x)+"' cy='"+str(midy)+"' r='"+str(2)+"' title='"+self.escape(taxon.name)+"' style='fill:black;'/>"
for p in self.id2project.values():
if p.taxon!=taxon:
continue
cx= width+ ((p.date - self.min_date)/(self.max_date - self.min_date))*width
cy= midy
print "<a target='ncbi' title='"+self.escape(taxon.name)+" projectId:"+str(p.id)+" size:"+str(p.genomeSize)+"' xlink:href='http://www.ncbi.nlm.nih.gov/sites/entrez?Db=genomeprj&cmd=ShowDetailView&TermToSearch="+str(p.id)+"'>"
print "<g style='stroke:red;' >"
if p.genomeSize!=None:
radius=max(2.0,((math.log10(p.genomeSize) - math.log10(self.min_size))/(math.log10(self.max_size) - math.log10(self.min_size)))*100.0)
print "<circle cx='"+str(cx)+"' cy='"+str(cy)+"' r='"+str(radius)+"' style='fill:orange;fill-opacity:0.1;'/>"
print "<line x1='"+str(cx-5)+"' y1='"+str(cy)+"' x2='"+str(cx+5)+"' y2='"+str(cy)+"'/>"
print "<line x1='"+str(cx)+"' y1='"+str(cy-5)+"' x2='"+str(cx)+"' y2='"+str(cy+5)+"'/>"
print "</g></a>"
for c in taxon.children:
total+= c.weight()
for c in taxon.children:
h2=height* (c.weight()/total)
y2=y+h2/2.0
w2=h_size
if len(c.children)==0:
w2=width-x
print "<polyline points='"+\
str(x)+","+str(midy)+" "+\
str(x+w2/2.0)+","+str(midy)+" "+\
str(x+w2/2.0)+","+str(y2)+" "+\
str(x+w2)+","+str(y2)+" "+\
"' title='"+self.escape(c.name)+"'/>"
self.recurs(c,x+w2,y,width,h2,w2)
y+=h2
class Taxon:
def __init__(self):
self.id=-1
self.name=None
self.parent=None
self.children=set()
def __hash__(self):
return self.id.__hash__()
def __eq__(self,other):
if other==None:
return False
return self.id==other.id
def weight(self):
w=1
for t in self.children:
w+= t.weight()
return w
def depth(self):
##sys.stderr.write(self.name+"\n")
d=0.0
for t in self.children:
d= max(t.depth(),d)
return 1.0+d
class Project:
def __init__(self):
self.id=-1
self.taxon=None
self.genomeSize=None
self.date=None
class GPHandler(handler.ContentHandler):
LIMIT=1E7
SLEEP=0
def __init__(self,owner):
self.owner=owner
self.content=None
self.project=None
def sql2date(self,s):
s=s[:s.index(' ')]
cal =time.mktime( time.strptime(s,"%m/%d/%Y") )
return cal
def startElementNS(self,name,qname,attrs):
if name[1]=="Document":
self.project=Project()
elif name[1] in ["ProjectID","TaxID","CreateDate","EstimatedGenomeSize"] :
self.content=""
def endElementNS(self,name,qname):
if name[1]=="Document":
if len(self.owner.id2project)<GPHandler.LIMIT:
self.owner.id2project[self.project.id]= self.project
self.project=None
elif name[1]=="TaxID":
if len(self.owner.id2project)<GPHandler.LIMIT:
self.project.taxon=self.owner.findTaxonById(int(self.content))
time.sleep(GPHandler.SLEEP) #don't be evil with NCBI
elif name[1]=="ProjectID":
self.project.id=int(self.content)
sys.stderr.write("Project id:"+self.content+" n="+str(len(self.owner.id2project))+"\n")
elif name[1]=="EstimatedGenomeSize" and len(self.content)>0:
self.project.genomeSize=float(self.content)
elif name[1]=="CreateDate":
self.project.date=self.sql2date(self.content)
self.content=None
def characters(self,content):
if self.content!=None:
self.content+=content
main=Main();
sys.stderr.write(main.escape("<>!")+"\n")
parser = make_parser()
# we want XML namespaces
parser.setFeature(handler.feature_namespaces,True)
# tell parser to disable validation
parser.setFeature(handler.feature_validation,False)
parser.setFeature(handler.feature_external_ges, False)
parser.setContentHandler(GPHandler(main))
parser.parse(sys.argv[1])
main.svg()
That's it,
Pierre

1 comment:
If you're going to do much with XML, you might want to look into the ElementTree module, which I find much nicer for reading, writing, and handling XML.
Post a Comment