Feeling like a newbie: Parsing NCBI-TinySeq with RUBY
I've been recently interested in the new popular language Ruby and its web framework Rails for two reasons:
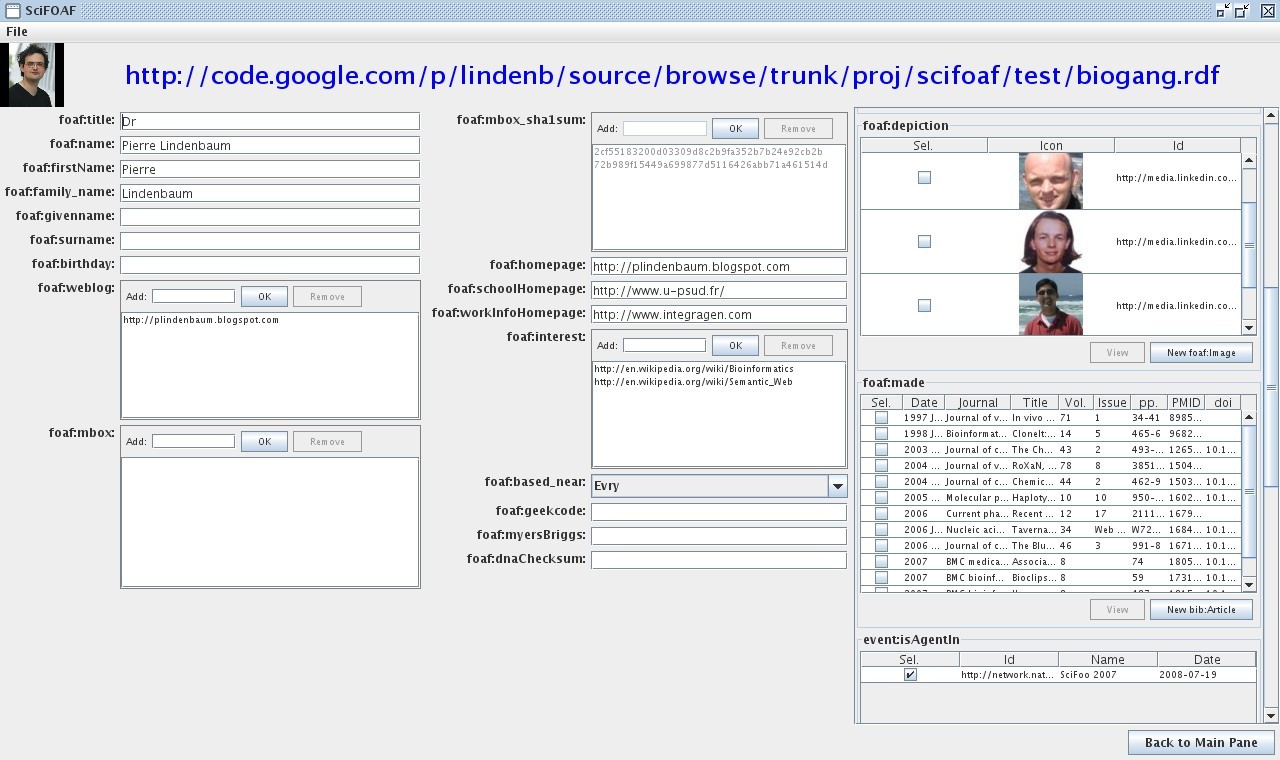
First,this picture:

Second, Matt Wood's blog about bioinformatics and ruby.
So here is my very first experience with ruby. The book I used was

Very good for learning but a little bit outdated. And I shoudl have started with a book about ruby only.
I've downloaded rails as described here. I got two problems:
- a ssl library was not installed. I fixed the problem thank to this post
- a problem with the zlib library, fixed by installing the ruby-zlib
My 'hello world' was "download a sequence from ncbi in TinySeq/XML format, parse the XML and create a new instance of 'a sequence' class". I've been suprised to find that ruby doesn't contain a decent API for parsing XML with DOM or SAX. The default API is called REXML and I found it ugly (or I may be too new to ruby to understand why it may be good). On friendfeed Adam Kraut suggested me to consider libxml-ruby (http://libxml.rubyforge.org/) or Hpricot (an xhtml parser). However I gave REXML a try by using its event-based API (but it is *not* a SAX API (namespaces are not supported) ).
OK, here is how I coded this (it took me hours to find the correct statements :-) ).
First we define a class handling an Organism which is just an id and a name (taxId and taxName)
class Organism
#constructor
def initialize(id,name)
@id=id
@name=name
end
end
#constructor
def initialize(id,name)
@id=id
@name=name
end
end
We also tell ruby to generate the getters to access those two properties
attr_accessor :id, :name
Next, we define a TinySeq class:
class TinySeq
#class members
@seqtype=nil
@gi=nil
@accver=nil
@sid=nil
@local=nil
@organism=nil
@defline=nil
@length=nil
end
#class members
@seqtype=nil
@gi=nil
@accver=nil
@sid=nil
@local=nil
@organism=nil
@defline=nil
@length=nil
end
In this TinySeq class I defined a nested class Handler: a XML stream handler using the REXML::StreamListener API. This looks like a SAX Handler but it doesn't handle the namespaces.
A TinySeq XML looks like this
<?xml version="1.0"?>
<!DOCTYPE TSeqSet PUBLIC "-//NCBI//NCBI TSeq/EN" "http://www.ncbi.nlm.nih.gov/dtd/NCBI_TSeq.dtd">
<TSeqSet>
<TSeq>
<TSeq_seqtype value="nucleotide"/>
<TSeq_gi>5</TSeq_gi>
<TSeq_accver>X60065.1</TSeq_accver>
<TSeq_taxid>9913</TSeq_taxid>
<TSeq_orgname>Bos taurus</TSeq_orgname>
<TSeq_defline>B.bovis beta-2-gpI mRNA for beta-2-glycoprotein I</TSeq_defline>
<TSeq_length>1136</TSeq_length>
<TSeq_sequence>CCAGCGCTCGTCT.....CAAGAAAAAAA</TSeq_sequence>
</TSeq>
</TSeqSet>
<!DOCTYPE TSeqSet PUBLIC "-//NCBI//NCBI TSeq/EN" "http://www.ncbi.nlm.nih.gov/dtd/NCBI_TSeq.dtd">
<TSeqSet>
<TSeq>
<TSeq_seqtype value="nucleotide"/>
<TSeq_gi>5</TSeq_gi>
<TSeq_accver>X60065.1</TSeq_accver>
<TSeq_taxid>9913</TSeq_taxid>
<TSeq_orgname>Bos taurus</TSeq_orgname>
<TSeq_defline>B.bovis beta-2-gpI mRNA for beta-2-glycoprotein I</TSeq_defline>
<TSeq_length>1136</TSeq_length>
<TSeq_sequence>CCAGCGCTCGTCT.....CAAGAAAAAAA</TSeq_sequence>
</TSeq>
</TSeqSet>
here is the code I used to handle the XML events:
class Handler
include REXML::StreamListener
....
include REXML::StreamListener
....
the class is initialized with an empty TinySeq instance
def initialize(tinyseq)
@tinyseq = tinyseq
#current tag
@tag = nil
#current text
@textcontent = nil
#did we see an error ?
@error=false
@taxid=nil
@taxname=nil
end
@tinyseq = tinyseq
#current tag
@tag = nil
#current text
@textcontent = nil
#did we see an error ?
@error=false
@taxid=nil
@taxname=nil
end
I wish I could have just declared some getters for the class TinySeq but this nested class TinySeq::Handler needed to set the properties of this tinyseq. It seems not to work like java where the nested classes have an access to the private properties of the parent class) that is why I asked ruby to create both setters and getters in TinySeq.
attr_accessor :seqtype, :gi, :accver, :sid, :local, :organism, :defline, :length
Next we define what the handler should do when it opens a tag
def tag_start(name, attrs)
@tag=nil
if !( name=="TSeq_sequence" || name=="TSeqSet" || name=="TSeq" || name=="TSeq_seqtype"|| name=="Error" )
@tag=name
@textcontent=""
elsif name=="TSeq_seqtype"
@tinyseq.seqtype = attrs["value"]
elsif name=="Error"
@error=true
end
end
@tag=nil
if !( name=="TSeq_sequence" || name=="TSeqSet" || name=="TSeq" || name=="TSeq_seqtype"|| name=="Error" )
@tag=name
@textcontent=""
elsif name=="TSeq_seqtype"
@tinyseq.seqtype = attrs["value"]
elsif name=="Error"
@error=true
end
end
... when it finds a text within a tag
def text(text)
unless @tag.nil?
@textcontent+=text
end
end
unless @tag.nil?
@textcontent+=text
end
end
... when it closes a tag (it fills the properties of the tinyseq)
def tag_end(name)
unless @tag.nil?
case @tag
when "TSeq_gi"
@tinyseq.gi = @textcontent
when "TSeq_accver"
@tinyseq.accver = @textcontent
when "TSeq_sid"
@tinyseq.sid = @textcontent
when "TSeq_local"
@tinyseq.local = @textcontent
when "TSeq_defline"
@tinyseq.defline = @textcontent
when "TSeq_taxid"
@taxid = @textcontent
when "TSeq_orgname"
@orgname = @textcontent
when "TSeq_length"
@tinyseq.length = @textcontent
else
$stderr.puts "Error #{name} #{@tag}\n"
end
end
if name=="TSeq" && !error
@tinyseq.organism= Organism.new(@taxid,@orgname)
end
@tag=nil
@textcontent=""
end
unless @tag.nil?
case @tag
when "TSeq_gi"
@tinyseq.gi = @textcontent
when "TSeq_accver"
@tinyseq.accver = @textcontent
when "TSeq_sid"
@tinyseq.sid = @textcontent
when "TSeq_local"
@tinyseq.local = @textcontent
when "TSeq_defline"
@tinyseq.defline = @textcontent
when "TSeq_taxid"
@taxid = @textcontent
when "TSeq_orgname"
@orgname = @textcontent
when "TSeq_length"
@tinyseq.length = @textcontent
else
$stderr.puts "Error #{name} #{@tag}\n"
end
end
if name=="TSeq" && !error
@tinyseq.organism= Organism.new(@taxid,@orgname)
end
@tag=nil
@textcontent=""
end
Then we create a static... sorry... a "class method" fetch for TinySeq returning a TinySeq object from a ncbi gi identifier.
def TinySeq.fetch(gi)
#build the efetch uri
#the API works whatever db is protein or nucleotide
url = "http://www.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=protein&id="+ CGI::escape(gi.to_s)+"&rettype=fasta&retmode=xml";
#create a new Streaming handler
handler= TinySeq::Handler.new TinySeq.new
#parse the document
REXML::Document.parse_stream(Net::HTTP.get_response(URI.parse(url)).body, handler)
if handler.error
return nil
end
return handler.tinyseq
end
#build the efetch uri
#the API works whatever db is protein or nucleotide
url = "http://www.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=protein&id="+ CGI::escape(gi.to_s)+"&rettype=fasta&retmode=xml";
#create a new Streaming handler
handler= TinySeq::Handler.new TinySeq.new
#parse the document
REXML::Document.parse_stream(Net::HTTP.get_response(URI.parse(url)).body, handler)
if handler.error
return nil
end
return handler.tinyseq
end
Finally, here is a good old 'main argc/argv' reading a list of gi on the command line, fetching a TinySeq object, putting this instance in an array and printing the content of the array.
seqarray=[]
ARGV.each{|gi|
seq=TinySeq.fetch(gi)
if seq.nil?
$stderr.print "#{gi} is a bad gi\n"
else
seqarray << seq
end
}
seqarray.each{|seq| print seq.to_s+"\n"}
ARGV.each{|gi|
seq=TinySeq.fetch(gi)
if seq.nil?
$stderr.print "#{gi} is a bad gi\n"
else
seqarray << seq
end
}
seqarray.each{|seq| print seq.to_s+"\n"}
... testing....
pierre@linux:~> ruby tinyseq.rb 5 6 7
(5|X60065.1) "B.bovis beta-2-gpI mRNA for beta-2-glycoprotein I" size:1136 (9913) Bos taurus
(6|CAA42669.1) "beta-2-glycoprotein I [Bos taurus]" size:342 (9913) Bos taurus
(7|X51700.1) "Bos taurus mRNA for bone Gla protein" size:437 (9913) Bos taurus
(5|X60065.1) "B.bovis beta-2-gpI mRNA for beta-2-glycoprotein I" size:1136 (9913) Bos taurus
(6|CAA42669.1) "beta-2-glycoprotein I [Bos taurus]" size:342 (9913) Bos taurus
(7|X51700.1) "Bos taurus mRNA for bone Gla protein" size:437 (9913) Bos taurus
That's it. I would be curious to know about a simpler and more elegant solution.
Pierre
The complete source code:
require "cgi"
require 'net/http'
require 'rexml/document'
require 'rexml/streamlistener'
#an organism: a taxon ncbi id and a name
class Organism
#generate the getters; getId() and getName()
attr_accessor :id, :name
#constructor
def initialize(id,name)
@id=id
@name=name
end
#toString method
def to_s
return "("+@id+") "+@name
end
end
#a ncbi TinySeq
class TinySeq
#class members
@seqtype=nil
@gi=nil
@accver=nil
@sid=nil
@local=nil
@organism=nil
@defline=nil
@length=nil
#generate the getters and the setters
#How can I just use attr_reader instead attr_accessor of and the nested class TinySeq::Handler have an access to those properties ?
attr_accessor :seqtype, :gi, :accver, :sid, :local, :organism, :defline, :length
#toString function
def to_s
return "(#{@gi}|#{@accver}) \"#{@defline}\" size:#{@length} #{@organism}"
end
#internal class
class Handler
include REXML::StreamListener
attr_reader :error, :tinyseq
#initialize with a tinyseq
def initialize(tinyseq)
@tinyseq = tinyseq
#current tag
@tag = nil
#current text
@textcontent = nil
#did we see an error
@error=false
@taxid=nil
@taxname=nil
end
def tag_start(name, attrs)
@tag=nil
if !( name=="TSeq_sequence" || name=="TSeqSet" || name=="TSeq" || name=="TSeq_seqtype"|| name=="Error" )
@tag=name
@textcontent=""
elsif name=="TSeq_seqtype"
@tinyseq.seqtype = attrs["value"]
elsif name=="Error"
@error=true
end
end
def tag_end(name)
unless @tag.nil?
case @tag
when "TSeq_gi"
@tinyseq.gi = @textcontent
when "TSeq_accver"
@tinyseq.accver = @textcontent
when "TSeq_sid"
@tinyseq.sid = @textcontent
when "TSeq_local"
@tinyseq.local = @textcontent
when "TSeq_defline"
@tinyseq.defline = @textcontent
when "TSeq_taxid"
@taxid = @textcontent
when "TSeq_orgname"
@orgname = @textcontent
when "TSeq_length"
@tinyseq.length = @textcontent
else
$stderr.puts "Error #{name} #{@tag}\n"
end
end
if name=="TSeq" && !error
@tinyseq.organism= Organism.new(@taxid,@orgname)
end
@tag=nil
@textcontent=""
end
def text(text)
unless @tag.nil?
@textcontent+=text
end
end
end
#class method (eq java static)
def TinySeq.fetch(gi)
#build the efetch uri
#the API works whatever db is protein or nucleotide
url = "http://www.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=protein&id="+ CGI::escape(gi.to_s)+"&rettype=fasta&retmode=xml";
#create a new Streaming handler
handler= TinySeq::Handler.new TinySeq.new
#parse the document
REXML::Document.parse_stream(Net::HTTP.get_response(URI.parse(url)).body, handler)
if handler.error
return nil
end
return handler.tinyseq
end
end
seqarray=[]
ARGV.each{|gi|
seq=TinySeq.fetch(gi)
if seq.nil?
$stderr.print "#{gi} is a bad gi\n"
else
seqarray << seq
end
}
seqarray.each{|seq| print seq.to_s+"\n"}

{kind=link}